INTRODUCTION
The importance of nutrition in the management of critically ill patients has been increasingly recognized over the years. Recent research has actively explored various aspects of nutrition in septic patients, such as the timing of oral nutrition initiation, routes of nutrient delivery, and appropriate methods of administration. Neurocritical care patients differ from other critically ill patients with medical conditions in several aspects that warrant special consideration. These differences stem from the unique neurovascular supply, metabolic characteristics of neurons, and specific clinical situations and treatments in neurocritical care.
The brain, accounting for only 3-5% of body weight, demands a disproportionately high energy supply, contributing to 20% of the total metabolic rate. This high metabolic demand is met by an elevated cerebral blood flow via neurovascular coupling. Neurons in the cerebral cortex are situated at the outermost region of the brain and receive blood supply not directly from large arteries but rather through low-pressure terminal vessels. In cases of increased intracranial pressure, neuronal damage in brain regions receiving low-pressure blood supply (e.g., cerebral cortex, deep basal nuclei) is accelerated.
When cerebral perfusion is not maintained due to increased intracranial pressure or hypotension, neurons may face energy supply disruptions even from astrocytes. This leads to intracellular energy deficiency, impairing various electrolyte pumps (e.g., Na-K pump) and neurotransmitter functions and accelerating secondary neuronal damage. Furthermore, neurons possess a unique signaling system in the form of neurotransmitters, requiring a continuous supply of amino acids as precursors. However, the type of amino acids needed varies depending on the clinical situation in neurocritical care patients, and excessive administration can provoke side effects such as seizures. Thus, it is crucial to understand and provide the appropriate amino acid supply according to the clinical context. Recent advances in nutritional support for neurocritical care patients, revealed amino acids play a critical role in various metabolic and neurotransmitter pathways in the brain. This review aims to explore the potential benefits of amino acid supplementation in neurocritical care, focusing on branched-chain amino acids (BCAAs), methionine, serine, arginine, glutamate, and selenium. Current research has provided some evidence for the potential role of these amino acids in enhancing hippocampal function, regulating brain function and energy production, promoting neuronal survival, and modulating vascular relaxation in patients with neurological injuries. However, the direct relationship between amino acid supplementation and patient outcomes remains inconclusive, highlighting the need for further research in this area.
Collectively, neurons exhibit both unique nutritional requirements and vulnerability, making proper nutrient supply essential. In neurocritical care, patients with elevated intracranial pressure or undergoing therapeutic hypothermia may have specific nutritional requirements. Therefore, it is necessary to revisit the energy requirements, metabolic characteristics, nutritional instability of neurons, and the impact of neurocritical care treatment techniques on nutritional demands in neurocritical care patients.
METHODS
To systematically narrative review the recent updates in decompressive craniectomy and cranioplasty, we conducted a comprehensive literature search using the following electronic databases: MEDLINE, EMBASE, and Cochrane Library. The search terms included "neurocritical care," "nutrition," “amino acid,” "intracranial pressure," "metabolism," "hypothermia," "traumatic brain injury," “indirect calorimeter,” and "stroke." The search was limited to articles published between January 2010 and December 2022, and only articles written in English were considered for inclusion.
Two independent reviewers screened the search results by title and abstract. Articles were considered for full-text review if they were deemed relevant to the study's objectives. Any disagreements between the reviewers were resolved by consensus or consultation with a third reviewer. Full-text articles were reviewed to determine eligibility for inclusion in the systematic review.
We included studies that reported on physiological changes following decompressive craniectomy, indications for decompressive craniectomy, optimal size of decompressive craniectomy, optimal timing of cranioplasty, syndrome of trephined, and the necessity of suboccipital cranioplasty. Both randomized controlled trials and observational studies were considered for inclusion. Case reports, case series, and expert opinions were excluded.
A narrative synthesis of the findings from the included studies was conducted, focusing on the physiological effects, indications, complications, and management of decompressive craniectomy and cranioplasty. Due to the heterogeneity in study design and reported outcomes, a meta-analysis was not performed. Instead, we present a descriptive summary of the available evidence, including a discussion of the strengths and limitations of the reviewed studies, and provide recommendations for clinical practice and future research.
LITERATURE REVIEW
The role of amino acid supplementation in neurocritical care
Amino acids, the fundamental components of proteins, consist of 20 different types. Although the body's proteins are incredibly diverse, encompassing hundreds of thousands of varieties, only 20 amino acids form their structure. These 20 amino acids are categorized into 9 essential amino acids (EAAs), 7 conditionally non-essential amino acids, and 4 non-essential amino acids. Essential amino acids cannot be synthesized within the body or are synthesized in inadequate amounts, necessitating their external supply to support physiological functions. Conditionally non-essential amino acids can be synthesized under normal circumstances, but their production may be impaired in specific situations, such as trauma, requiring an external supply. Non-essential amino acids are easily synthesized within the body.
Utilization of branched-chain amino acids in neurocritical care
Branched-chain amino acids (BCAAs), including valine, leucine, and isoleucine, have a tree branch-like structure, are hydrophobic, and promote protein synthesis (Fig. 1). BCAAs serve as precursors for the neurotransmitters glutamate and GABA (gamma-aminobutyric acid) and are essential components of the energy metabolism-related citric acid cycle (Krebs cycle)1).Studies in animal models and clinical settings have confirmed decreased BCAA concentrations in the hippocampus and plasma following traumatic brain injury (TBI)2,3).This observation has led to the hypothesis that BCAA supplementation after TBI may enhance hippocampal function by providing a source for the synthesis of glutamate and GABA.
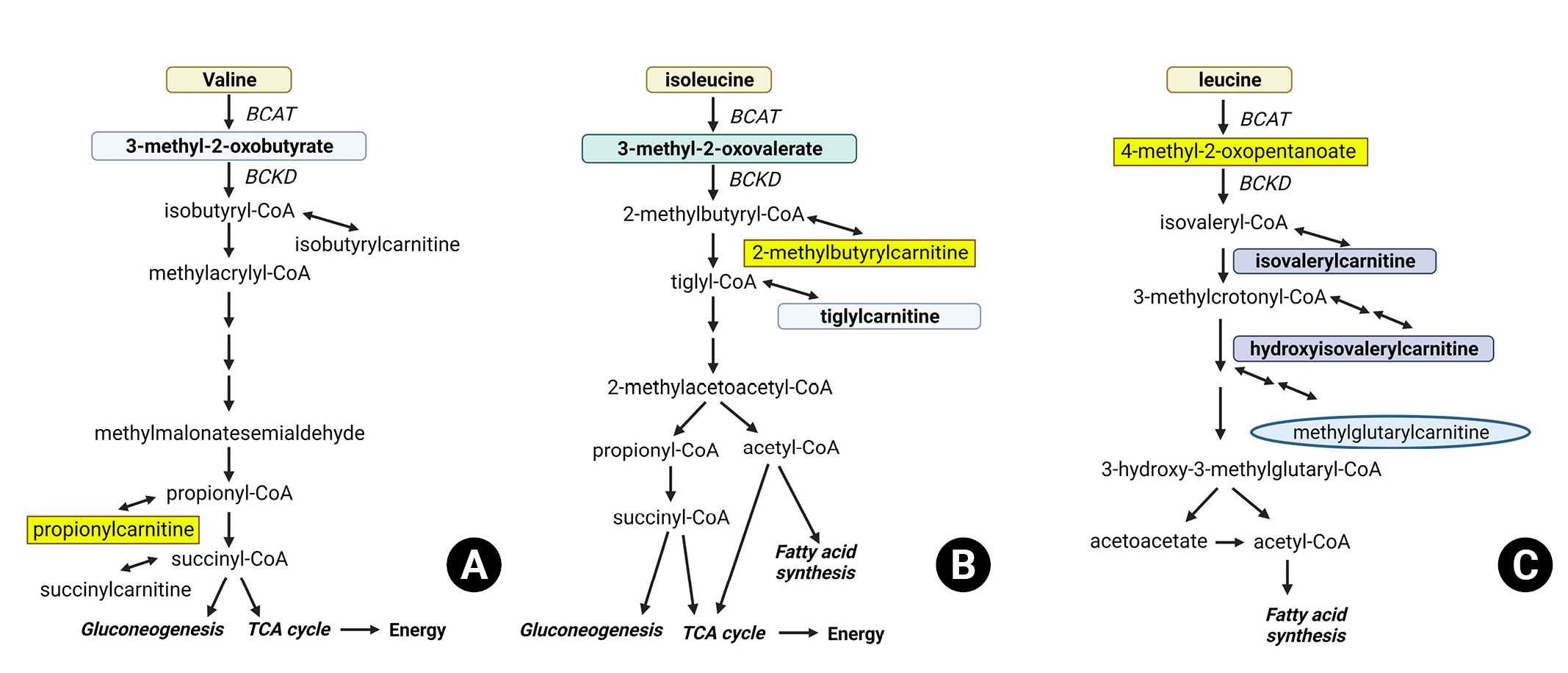
In a mouse model of brain injury, a decrease in hippocampal BCAA concentrations was observed. Subsequent BCAA supplementation in the brain-injured subjects improved their conditioned fear responses to levels similar to those of the control group, while simultaneously increasing hippocampal BCAA concentrations4). Additionally, BCAA supplementation was advantageous in maintaining wakefulness. A study involving patients with TBI revealed significant decreases in BCAA and metabolite concentrations3). Plasma BCAA concentrations decreased following brain injury, with more severe injuries showing more significant reductions. These findings suggest that changes in BCAA metabolism after TBI may affect the pathophysiology of TBI by causing insufficient energy production and neurotransmitter synthesis. Researchers have found considerable associations between decreased blood concentrations of BCAA metabolites, such as propionylcarnitine, 2-methylbutyrylcarnitine, and 4-methyl-2-oxopentanoate, and increased intracranial pressure (Fig. 2).
The exact cause of reduced BCAA metabolism in traumatic brain injury remains unclear. However, it could be related to metabolic disorders of enzymes involved in BCAA metabolism, such as reversible transamination by branched-chain aminotransferase and irreversible dehydrogenation by branched-chain α-keto acid dehydrogenase. These enzymes play an inevitable role in the initial stages of BCAA metabolism, and their dysfunction may be linked to the observed decrease in BCAA metabolism following injury5).
The association between bcaas levels and altered consciousness in neurocritical care patient
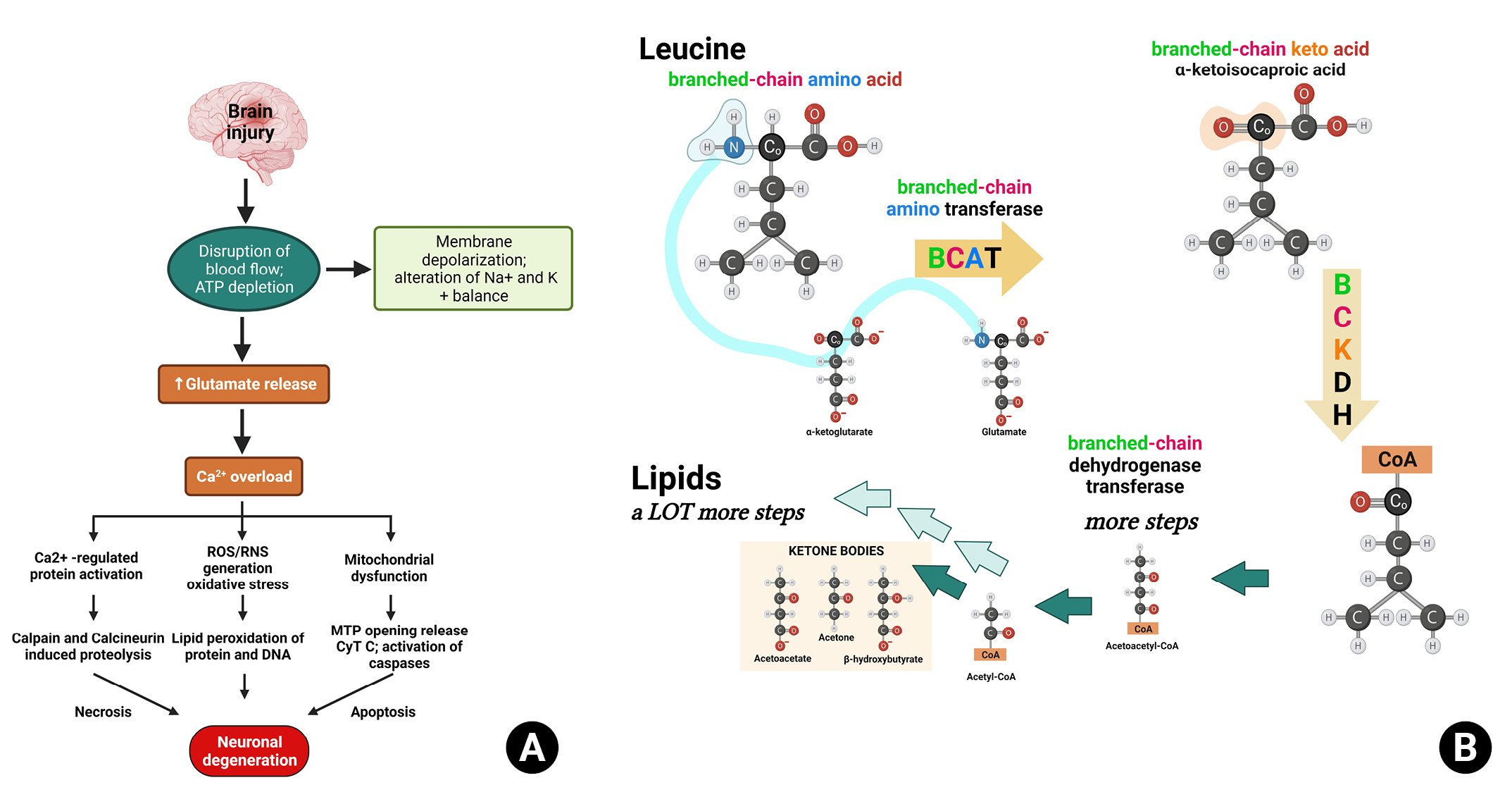
Although a definitive conclusion about the direct relationship between serum BCAA concentrations and brain function remains elusive, there is evidence supporting this possibility. First, the glucogenic energy production of valine and isoleucine, as well as the ketogenic energy production of leucine and isoleucine, play a role in this connection6). BCAAs pass through the blood-brain barrier using energy-dependent transporters. In injured brains, blood flow alterations can lead to ATP deficiency (Fig. 3A). Consequently, leucine generates energy through the ketogenic pathway, replacing glucose in the brain, and increasing the secretion of its metabolic product, glutamate (Fig. 3B). Thus, the use of BCAAs as an energy source accelerates TBI-mediated glutamate excitotoxicity, leading to secondary neuronal damage (Fig. 3A).
The second line of evidence highlights the amino acid imbalance resulting from decreased BCAAs. In patients with long-standing brain injuries, serum BCAA levels decrease even further. Contrary to the previous discussion, a prolonged state of reduced leucine may lead to a decrease in excitatory neurotransmitter glutamate, resulting in diminished synaptic activity and neuroplasticity, ultimately causing cognitive and behavioral disorders1). Moreover, when BCAA serum concentrations decrease, large neutral amino acids (LNAAs, including tryptophan, tyrosine, and phenylalanine) are utilized instead. This substitution can increase serotonin and catecholamine concentrations, potentially leading to behavioral and cognitive abnormalities, such as bipolar disorder and schizophrenia, when present in excess7,8).
Based on this evidence, the external supply of BCAAs has been proposed as a potential therapeutic approach. In fact, BCAA administration in patients with severe traumatic brain injury (TBI) has significantly aided cognitive recovery9). Other studies have reported that maintaining normal protein and caloric intake in patients with TBI may enhance cognitive recovery through nutritional BCAA supplementation10).
The role of methionine and serine in cognitive function of neurocritical care patients
1) Methionine
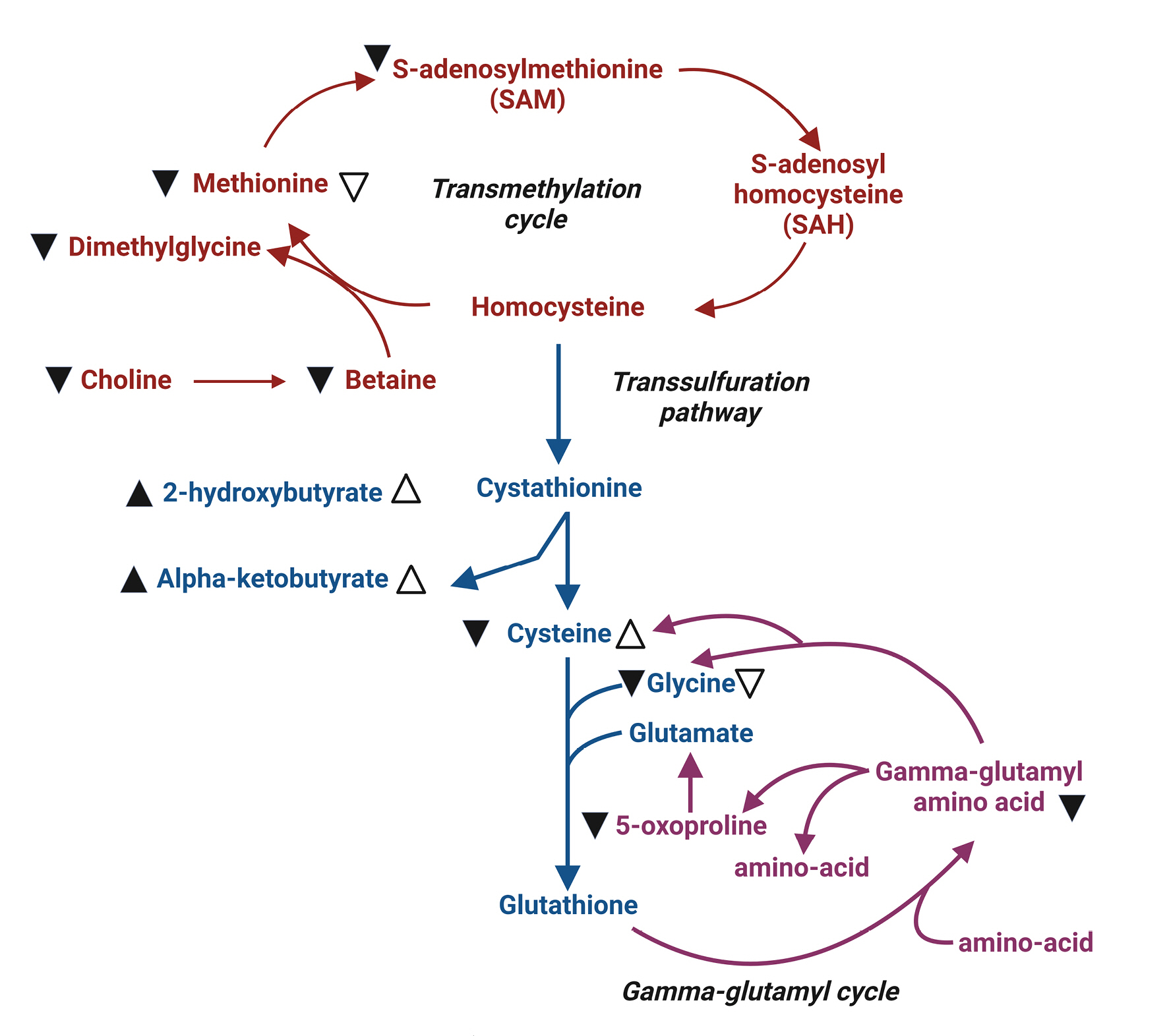
Methionine serves as a methyl group donor during methylation. Its metabolite, S-adenosylmethionine (SAM), plays a crucial role in gene expression and cellular signaling (Fig. 4). Additionally, methionine is involved in the synthesis of glutathione, which protects cells from oxidative stress. The brain, with its high lipid content and metabolic activity, is particularly vulnerable to oxidative stress. Consequently, a decrease in glutathione levels due to brain injury may accelerate brain damage11). In cases of severe traumatic brain injury (TBI), methionine, SAM, and glutathione levels decline, leading to various cellular changes. These changes include reductions in protein synthesis, gene expression, and cellular protection via glutathione production. In fact, decreased serum concentrations of methionine, BCAAs, and histidine have been reported in patients with severe TBI12). However, no direct evidence currently supports the external supplementation of methionine to improve the prognosis of patients with severe TBI.
2) Serine
Serine is a non-essential amino acid that can be synthesized in the body using glycine as a precursor. Serine is metabolized into phosphatidylserine (PS). Serine crosses the blood-brain barrier via sodium-dependent neutral amino acid transporters and is present in cerebrospinal fluid at approximately 10% of plasma concentration. In neurons, serine is metabolized to PS. Neuron-derived PS is rich in docosahexaenoic acid (DHA), and the supply of DHA promotes the synthesis of PS. PS is a major phospholipid in the inner layer of cell membranes in neural tissues, accounting for 13-15% of the phospholipids in the cerebral cortex. Located in the neuronal cell membrane, PS regulates synaptic receptor expression and neurotransmitter release, thus participating in signal transduction related to neuronal survival, neurite growth, and synaptic genesis13).
The clinical significance of serine supplementation is still under investigation. Ethanol has been shown in animal studies to reduce DHA levels and degrade PS in the hippocampus, impairing neuronal survival and function and causing apoptosis of hippocampal cells14,15). Patients with Alzheimer's dementia, who exhibit memory and cognitive decline, have been found to have decreased DHA and PS levels. Oral administration of serine in the form of PS has been reported to improve cognitive and verbal call functions in Alzheimer's patients. However, more research is needed to establish the potential benefits of serine supplementation in neurocritical care patients16,17).
The role of arginine in cerebrovascular constriction and vasospasm
Arginine serves as a precursor for nitric oxide (NO), which functions as an endothelium-derived relaxing factor. NO is produced from arginine through the action of endothelial NO synthase (eNOS) in the cerebral endothelium and neuronal NOS (nNOS) in the adventitia. NO production is stimulated in response to shear stress, metabolic demands, and chemoregulation, leading to vasodilation18).
In the context of subarachnoid hemorrhage (SAH), hemoglobin released into the subarachnoid space destroys nNOS-contacting neurons, inhibiting NO synthesis and leading to cerebrovascular constriction. When vascular constriction occurs, eNOS activation induced by shear stress is counteracted by the activation of endogenous competitive NOS inhibitors such as asymmetric dimethylarginine (ADMA). During cerebrovascular constriction, the enzyme responsible for ADMA removal (dimethylarginine-dimethylaminohydrolase II, or DDAH II) is unable to function properly due to an immune response, resulting in increased cerebrospinal fluid ADMA levels and exacerbating cerebrovascular constriction18) . Consequently, externally supplied arginine has been proposed as a potential treatment for cerebrovascular constriction by stimulating DDAH II and inhibiting L-arginine methylating enzyme19).
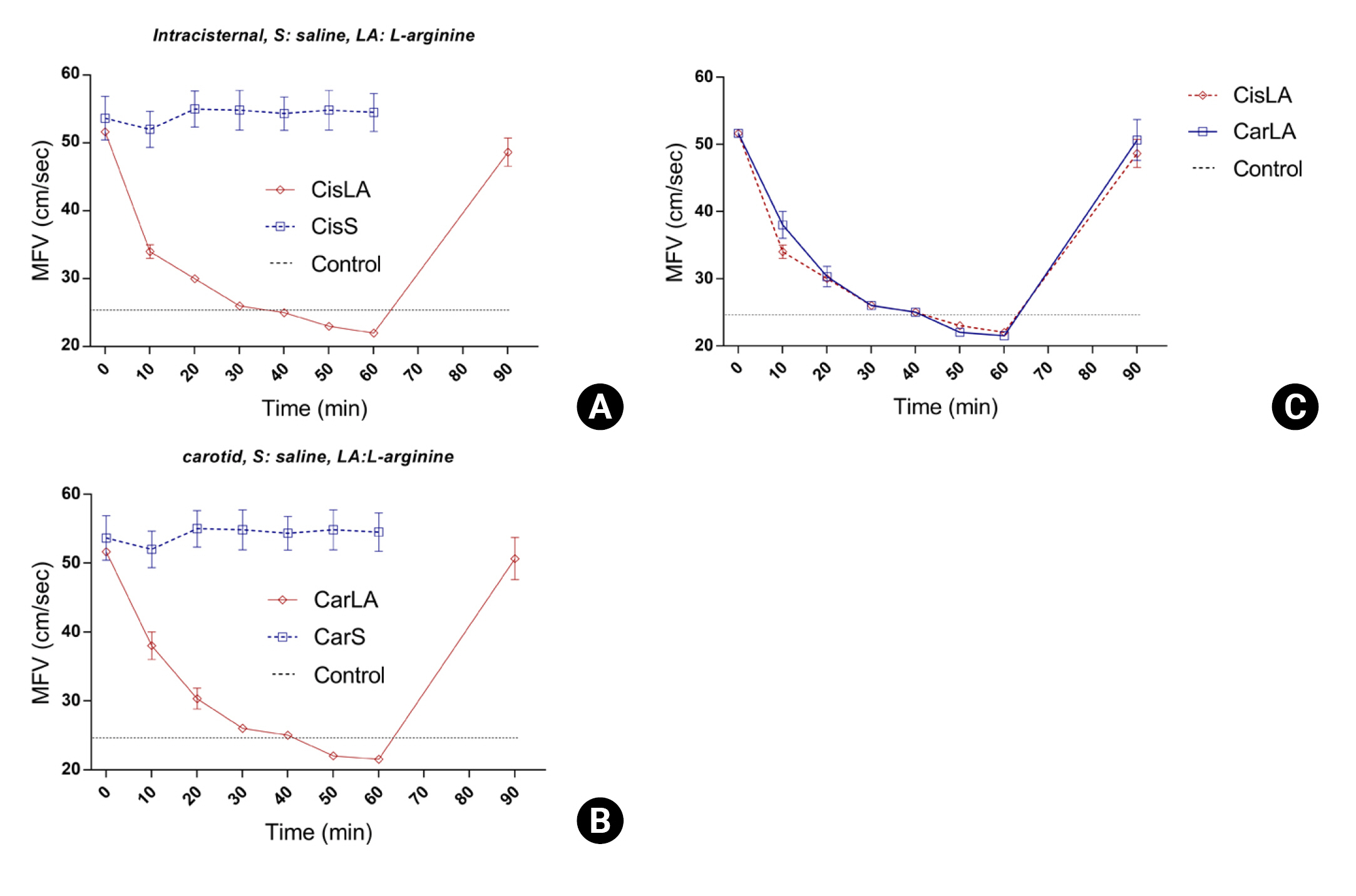
Animal studies have demonstrated that the administration of L-arginine ameliorates cerebrovascular constriction. When administered intracarotidly, the degree of improvement in cerebrovascular constriction was assessed using transcranial Doppler (TCD) to evaluate mean flow velocity (Fig. 5)20).In another study, an SAH vasospasm animal model showed that the administration of L-arginine, compared to saline, reduced the latency of motor evoked potentials and increased amplitude, resulting in improved clinical symptoms21). Another group directly observed changes in the vascular wall due to vasospasm and arginine administration using rat femoral artery samples19). Compared to the control group, the vasospastic vessels displayed increased wall thickness and non-intact endothelium. In the experimental group treated with L-arginine following vasospasm, an improvement in wall thickness and thinning was observed. These findings suggest that arginine administration may represent a novel therapeutic approach to treating cerebrovascular constriction in the context of subarachnoid hemorrhage.
Glutamate: to feed or not?
Glutamate is the most abundant free amino acid in the brain, and it serves as a neurotransmitter that transmits excitatory signals, is oxidized for energy production, and acts as a precursor for the synthesis of proteins, glutamine, GABA, and glutathione. Furthermore, it participates in cellular removal processes, thus playing a role in neural plasticity. Glutamate receptors are located on the surface of nerve cells, and excessive excitatory signals from glutamate can lead to cell death through a phenomenon called 'excitotoxicity.' Therefore, it is crucial for glutamate to be present in the appropriate concentration, location, and timing. A balance between glutamate transporter, which removes extracellular glutamate, and the blood-brain barrier, which prevents the movement of glutamate from the blood to the brain, is essential22).
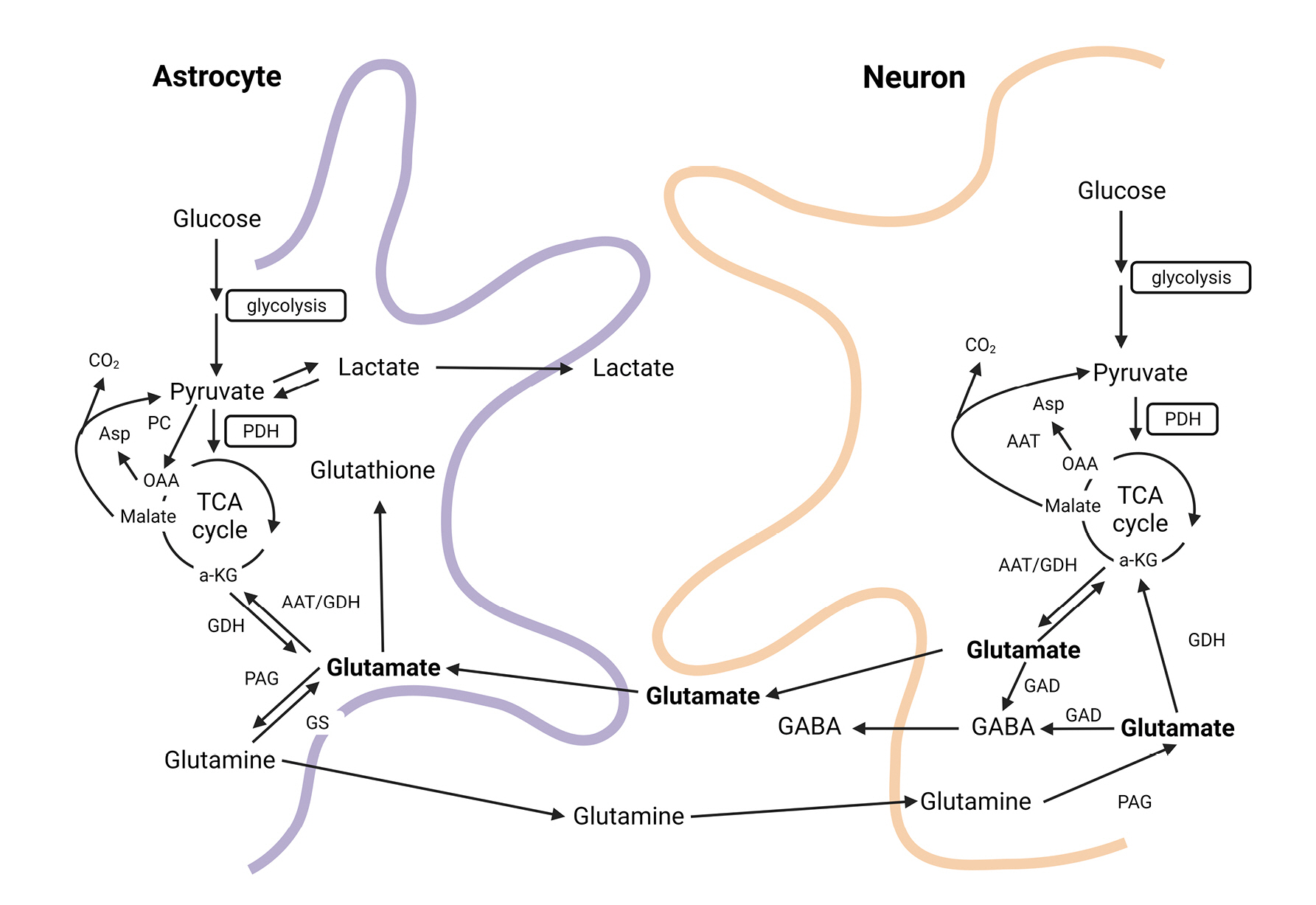
Glutamine is the most abundant amino acid in the human body and is primarily produced in muscles. It is a conditionally essential amino acid consumed in greater quantities during severe illness or following major surgery. Glutamine is responsible for more metabolic functions in the body than other amino acids. It can be converted into glucose to provide energy when needed, serves as a backbone for RNA/DNA synthesis, participates in ammonium production in the kidneys for acid-base balance, and is involved in the synthesis of the antioxidant glutathione23-25). In the central nervous system, glutamine acts as a precursor for neurotransmitters such as glutamate, aspartate, and GABA, and is synthesized in astrocytes through the glutamate-glutamine cycle. The synthesized glutamine is then transported to neurons and recycled as a precursor for glutamate and GABA (Fig. 6)26,27).
One of the central nervous system disorders characterized by an imbalance in the glutamate-glutamine cycle is epilepsy. An imbalance in glutamatergic and GABA-ergic transmission results in insufficient glutamine synthesis, leading to a lack of GABA-ergic activity and hyperexcitation, which manifests clinically as epilepsy. External glutamine supply can reverse this process. However, since glutamine is metabolized in the liver, liver dysfunction or excessive glutamine administration can lead to brain edema, increased intracranial pressure, and neurological abnormalities. Therefore, liver function should be monitored concurrently with glutamine administration22,23).
Glutamine is also involved in glutathione synthesis, protecting cells from oxidative stress, and a deficiency in glutamatergic neurotransmission may lead to emotional disorders and depression28). In animal models of stress-induced cognitive impairment, glutamine supplementation has shown significant positive results in recognition and location memory tests, suggesting its potential usefulness in preclinical settings29). In humans, increased mortality has been observed in critically ill patients with low blood glutamine levels, leading to ASPEN and ESPEN guidelines recommending glutamine administration with a level of evidence30,31). In cases of traumatic brain injury with blood-brain barrier damage, an appropriate external glutamine supply that does not increase glutamate concentration should be established to prevent exacerbation of excitotoxicity and brain edema32, 33). Although a study in severe traumatic brain injury patients administering 0.34g/kg of glutamine within 20 hours showed a significant increase in glutamine levels without elevating glutamate levels, the clinical outcomes remain unclear, necessitating further research34).
Considering this, it is advisable for each hospital to limit the administration of glutamate during the initial stages of brain injury when using amino acid preparations or total parenteral nutrition. This is due to the potential for intravenous nutrition containing glutamate to exacerbate brain injury during periods of rapid progression and uncontrolled brain edema caused by damage to the blood-brain barrier. Once hemodynamically stable and brain edema begins to be controlled, it is recommended to initiate enteral feeding and supplement with parenteral nutrition as needed.
Selenium: a beneficial element in neurocritical care?
Selenium exists in the human body in the form of 25 different selenoproteins and serves various functions. It is involved in numerous neural signal transmissions through neurotransmission. Selenium plays a role in maintaining motor function, coordination, memory, and cognition, and its deficiency can manifest as symptoms of Alzheimer's disease, Parkinson's disease, and epilepsy. In the cerebral cortex and hippocampus, it serves a GABA-ergic function, leading to refractory seizures in children when deficient. In the dopamine pathway, it exhibits both neurotoxic and neuroprotective properties depending on its concentration. It also participates in acetylcholine neurotransmission, contributing to the phosphorylation of antioxidant proteins and the maintenance of ion channel/calcium homeostasis at the molecular and cellular levels35).
Under stress conditions, the stress hormone glucocorticoid affects the cerebral cortex and hippocampus, leading to memory and emotional disorders. Glucocorticoids suppress the expression of selenoprotein genes, but the administration of exogenous selenium can counteract this effect and inhibit oxidative damage (lipid peroxidation) caused by glucocorticoids36,37). Animal studies have shown that selenium supplementation enhances hippocampal neurogenesis by mediating the proliferation of neuronal precursor cells in response to physical activity stimulation, thereby improving cognitive decline associated with hippocampal injury and aging38). In actual traumatic brain injury patients, intravenous administration of selenium for up to 10 days (1000mcg q.d. for 5 days followed by 500mcg q.d. for the next 5 days) did not show a significant difference in mortality rates. However, functional outcomes at discharge and 6-month follow-up were significantly improved in the patient group receiving selenium39). In patients with non-abdominal trauma, supplementation of selenium resulted in significant reductions in mortality, ICU, and overall hospital stays36). A meta-analysis of critically ill patients did not yield significant positive results for overall hospital stays, pneumonia, or side effects of renal failure in the selenium treatment group. Although a significant statistical value was obtained for mortality, this was on the borderline40). Studies on the clinical outcomes of selenium supplementation in patients with systemic inflammatory response syndrome (SIRS) and sepsis have yielded inconsistent results41). Although the effects of selenium supplementation are not consistent, it is considered "not inferior" and warrants weak recommendation for administration.
Special considerations in nutritional support for neuro-critical patients
Changes in energy requirements in neuro-critical patients depending on clinical situations
There are several factors to consider in providing appropriate energy supply to neuro-critical patients. First, the brain accounts for a significant portion of the body's total energy requirements, so it is essential to consider metabolic changes in the brain due to brain injury. Second, brain metabolism varies depending on the cause of the brain injury (especially trauma, subarachnoid hemorrhage due to aneurysm rupture, and cerebral infarction), hormonal changes, accompanying injuries, sepsis, pneumonia, and inflammatory responses due to complications. Third, many treatments used in the management of neuro-critical patients, such as sedatives, muscle relaxants, and hypothermia, can induce changes in energy metabolism.
1) Increased intracranial pressure
Animal experiments have shown that anaerobic glycolysis occurs in the brain during increased intracranial pressure, leading to decreased pH, ATP, and phosphocreatine. Energy metabolism in the brain is preserved until cerebral perfusion pressure drops below 30 mmHg42). When intracranial pressure rises, immediate changes in energy metabolism appear in the brain, and glucose, lactate, and pyruvate in the extracellular fluid decrease rapidly43). This indicates that energy production becomes insufficient due to increased intracranial pressure. Conversely, when intracranial pressure is reduced rapidly, glucose, lactate, and pyruvate return to normal levels42,43).
Although the degree of metabolic increase after brain injury is known to correlate with intracranial pressure, treatments for elevated intracranial pressure can also affect overall metabolic rate44). Since treatment aims to reduce intracranial pressure rapidly and avoid sustained elevated intracranial pressure, it is not easy to predict changes in energy requirements due to intracranial pressure fluctuations. Despite the theoretical expectation that thiopental should decrease CMRO2 and thus reduce additional energy consumption, studies have shown that thiopental does not reduce additional energy consumption compared to fentanyl and midazolam sedation. This suggests that efforts to reduce brain metabolism may not be effective45,46).
2) Trauma
It is well known that trauma triggers hypermetabolism and a catabolic state. In patients with head trauma, the metabolic rate is reported to increase by approximately 100-160%, although the extent varies in the literature47). In a study involving patients with traumatic brain injury, the basal metabolic rate was elevated compared to healthy individuals, with rates of 168±53% for those with a Glasgow Coma Scale (GCS) score of 5 or below, 129±31% for scores of 6-7, and 150±49% for scores of 8 or above. The resting metabolic rate (RMR) during the stable phase increased by 45% for each 1-degree increase in body temperature in patients with GCS scores of 5 or below and by 15% in those with scores of 6-7. In patients with GCS scores of 8 or above, no correlation was found between RMR and body temperature48). Considering that patients with GCS scores of 5 or below showed posturing responses to pain and persistent rigid muscle tone, and those with GCS scores of 8 or above exhibited more agitation, the lowest basal rate observed in patients with GCS scores of 6-7 may suggest that muscle contraction has a greater influence than the severity of brain injury. In patients with traumatic brain injury, the energy metabolic rate is typically 120-250% higher than the basal energy expenditure calculated using the Harris-Benedict equation, and it ranges from 76-120% when sedatives, paralytics, or barbiturates are used. Therefore, providing 140% of the predicted basal energy expenditure (BEE) is necessary for patients without paralysis49).
3) Stroke
The energy requirements for stroke patients have been reported variably in the literature. Stroke patients can be broadly categorized into ischemic stroke and hemorrhagic stroke.
In acute ischemic stroke patients, the total energy expenditure (TEE) is lower compared to other critically ill patients. When providing caloric supply to typical critically ill patients, there is a higher risk of overnutrition. The Harrison-Benedict equation (HBE) shows a relatively high correlation with the predicted TEE, so it is recommended to use HBE or indirect calorimetry to evaluate nutritional requirements50).
In contrast, high-weight based energy calculations (30kcal/kg) are better predictors of resting energy expenditure (REE) in hemorrhagic stroke patients, while low-weight based energy calculations (25kcal/kg) are better predictors in acute ischemic stroke patients51). Hemorrhagic stroke patients had a basal energy expenditure (BEE) of 126% (101-170%) during the first week, which was not statistically different from the mean BEE of 147% (114-176%) in patients with severe traumatic brain injury. Therefore, similar to patients with traumatic brain injury, hemorrhagic stroke patients have an increased metabolic rate, and there is a risk of under-supplying nutrition compared to standard critically ill patients52). In a study of spontaneous intracerebral hemorrhage patients, the average REE increased by 117.5%, and the energy requirement peaked between 7-10 days as time progressed. In patients with aneurysmal subarachnoid hemorrhage, the energy requirement initially started close to 25kcal/kg but gradually increased, reaching above 30kcal/kg on the sixth day53).
Influence of sedation and neuromuscular blockade on energy demand in neurocritical care patients
Many medications used in neurocritical care, particularly sedatives and muscle relaxants, have an impact on energy requirements. Although the reported effects vary in the literature, sedation with midazolam and fentanyl has been shown to decrease the average resting metabolic rate (RMR) by 6–33%, while neuromuscular blockers reduce it by 11–33%54-57). No significant difference in metabolic rate reduction was observed between propofol and midazolam54), with some studies showing a decrease in energy requirements by an average of 25%.
Fever has been reported to increase energy consumption by 10% per 1℃, and sepsis raises energy consumption regardless of fever46). In sedated brain-injured patients, sepsis and body temperature have been reported as the main causes of changes in energy consumption. The relationship between sedation and energy consumption suggests that the deeper the sedation, the lower the energy consumption55,56). When sedation is deep enough to eliminate spontaneous movement, there seems to be little difference between drugs.
1) Propofol
Propofol is a widely used sedative in neurocritical care patients due to its effect on reducing intracranial pressure. Propofol is formulated as a 10% lipid emulsion of soybean oil, which itself provides 11 kcal/g (1.1 kcal/mL) of energy. If this is not considered, it can lead to an oversupply of calories, hypertriglyceridemia, and inappropriate protein provision57). When using commercial intravenous nutrition products simultaneously, lipid-free total parenteral nutrition (TPN) can be used, or if a fat-containing multi (3)-chamber bag is used, the fat-containing seal should not be opened, and only the catheter port of the dextrose and amino acid-containing chamber bag should be connected, allowing the administration of a lipid-free dextrose and amino acid solution.
Changes in energy requirements during targeted temperature management (ttm) in neurocritical care patients
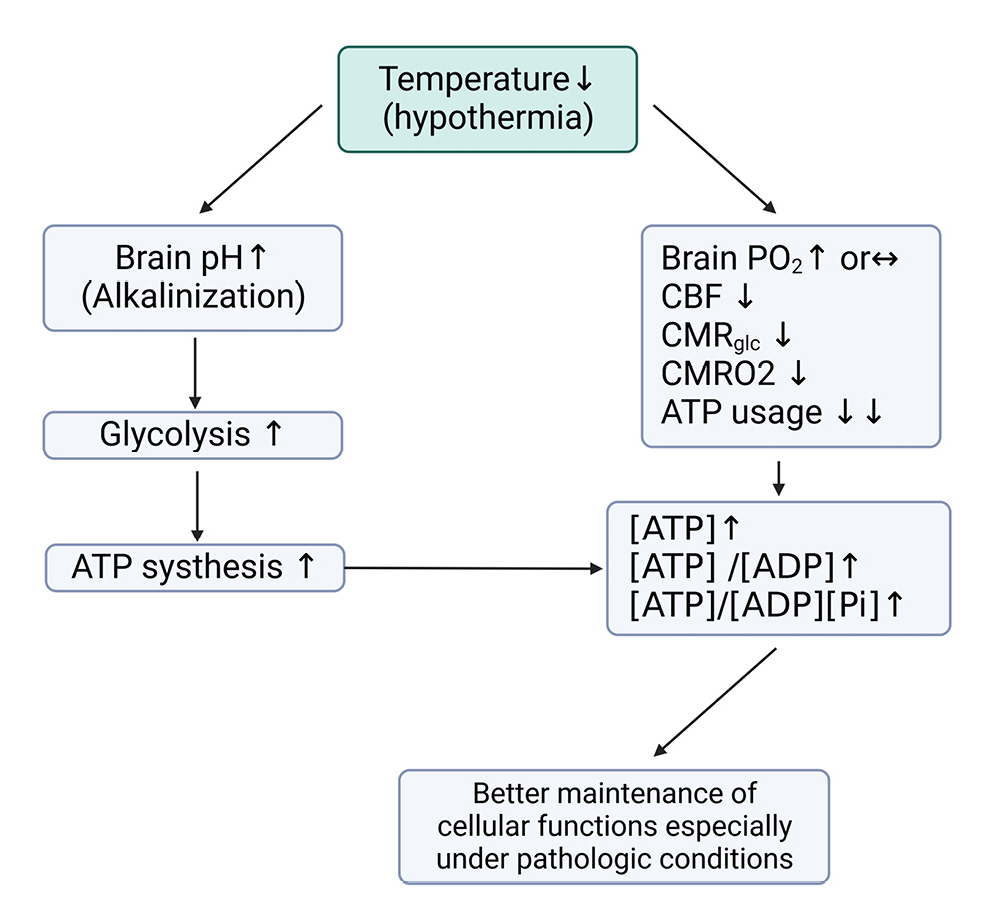
Changes in body temperature are one of the main factors affecting energy requirements, with a 1℃ change in temperature causing a 5-7% decrease in cerebral metabolic rate58) and a 10-13% change in energy requirements46). Theoretically, when reducing the body temperature from 37℃ to 33℃ during TTM, at least a 40% reduction in energy requirements should occur, but in reality, a 20–30% reduction has been observed (Fig. 7)59-61). This difference is attributed to hypermetabolic states caused by shivering, infection, and trauma, which make accurate prediction of energy requirements challenging.
According to the POLAR-RCT study, which compared patients undergoing prophylactic TTM at 33℃ for 3–7 days with traumatic brain injury patients maintaining a normal body temperature of 37℃, the average energy requirement on day 3 was 21 kcal/kg in the TTM group, a 20% reduction compared to the 27 kcal/kg in the normal body temperature group. On day 7, the TTM group's requirement was 25 kcal/kg, a 12% decrease compared to the normal group's 28 kcal/kg. After TTM, between days 8–14, a hypermetabolic state was confirmed with 33 kcal/kg61). In patients with cerebral infarction, energy requirements decreased by 29% during TTM at 33℃ and increased to 116% after rewarming59).
Nutritional support in patients with targeted temperature management (ttm)
While early enteral nutrition is recommended for both general critically ill patients and those with severe head injuries, the appropriate nutritional support method for neurocritical care patients undergoing TTM remains unclear. Enteral nutrition should be approached with caution during TTM below 34℃ due to the potential for paralytic ileus caused by reduced bowel motility and hemodynamic instability. It is often recommended to delay enteral nutrition until rewarming due to the risk of bowel ischemia or necrosis. ESICM recommends starting low-dose enteral nutrition early and increasing the supply after rewarming, although the evidence level is low (Grade 2D)62,63).
Studies on the safety of enteral nutrition during TTM exist, and research results indicate that early enteral nutrition is safe during TTM at 33-34℃ in cardiac arrest and hypoxic-ischemic encephalopathy patient groups60,61,63-67). An RCT study conducted in patients with traumatic brain injury showed that although the TTM group experienced more interruptions in enteral nutrition due to increased gastric residual volume, the reduced energy requirements due to TTM enabled them to better meet their energy needs compared to the normothermic group61). A study in patients with intracerebral hemorrhage found that enteral nutrition was delayed in TTM patients compared to the normothermic group, and the average caloric supply from days 0-3 was lower (398 kcal compared to 1006 kcal in the normothermic group). However, there was no association with adverse GI-related events or the occurrence of ventilator-associated pneumonia (VAP), indicating the feasibility of early enteral nutrition in TTM patients64). These studies are limited by small patient numbers and a focus on safety and meeting nutritional requirements during TTM in neurocritical care patients; no research has been conducted on improving outcomes, so caution is needed when interpreting the impact of nutrition on neurological outcomes.
Nevertheless, the benefits of enteral nutrition, including the preservation of intestinal mucosal function and maintenance of immune function, advocate for the implementation of early enteral nutrition even during targeted temperature management. It is recommended to monitor for gastrointestinal intolerance and adjust the dosage based on the patient's clinical presentation.
CONCLUSION
In conclusion, optimizing energy supply and metabolic support in neuro-critically ill patients requires a detailed understanding of the specific roles and interactions of various amino acids and micronutrients, such as branched-chain amino acids (BCAAs), methionine, serine, arginine, glutamate, and selenium. These elements are crucial in modulating neuronal function, energy metabolism, and neuroprotection, ultimately influencing patient outcomes.
BCAAs, comprising valine, leucine, and isoleucine, are precursors to the neurotransmitters glutamate and GABA, and serve as key components of the citric acid cycle. Methionine is essential for the synthesis of s-adenosylmethionine (SAM) and glutathione, both of which play pivotal roles in gene expression, cellular signaling, and protection from oxidative stress. Serine contributes to the formation of phosphatidylserine (PS), a crucial component of neuronal cell membranes that regulates synaptic receptor expression and neurotransmitter release. Arginine, a precursor to nitric oxide (NO), is involved in vascular relaxation and has been suggested as a potential therapeutic target for vascular constriction.
Understanding the complex relationship between BCAA levels and altered brain function, as well as the potential benefits of supplementing other amino acids and micronutrients, is imperative for tailoring nutritional interventions for neuro-critical patients. Furthermore, special considerations in nutritional support encompass the assessment of energy demands, which may be influenced by the severity and etiology of brain injury, concomitant drug therapy (including sedatives and neuromuscular blockers), and targeted temperature management.
Future research should focus on elucidating the intricate relationships between these nutritional factors and their impact on neuro-critical patients, with the aim of establishing evidence-based guidelines to enhance metabolic support. By refining our understanding of these elements and tailoring nutritional interventions, we can ultimately contribute to improved patient outcomes and expedite recovery in this vulnerable population.