INTRODUCTION
Thrombotic thrombocytopenic purpura (TTP) is a multi-systemic microvascular disorder caused by imbalance between the formation of unusually large von Willebrand factor (vWF) multimer and cleaving proteinase ADAMTS131,2) TTP is used to present a constellation of symptoms of microangiopathic hemolytic anemia (MAHA), characteristic peripheral blood smear findings, fever, renal failure and neurologic abnormalities3). It is sometimes characterized by combination of atypical clinical features, make it difficult to differentiate from other diseases such as idiopathic thrombocytopenic purpura (ITP), disseminated intravascular coagulation (DIC) and sepsis. Specifically, the diagnosis is challenging when TTP is maninfestated with multifocal cerebral infarction because many clinical conditions could result in. With the development of recent molecular diagnostics based on the ADAMTS 13 assay, the diagnosis rate of TTP has been refined compared to the past. However, the point that needs to be careful in the diagnosis of TTP is to consider the low sensitivity of ADAMTS 13 assay although it showed high specificity3,4). Therefore, in the case of clinical presumptive TTP, the ADAMTS 13 assay is often demonstrated negative results. Although it is certain that the ADAMTS 13 assay is an important molecular method for diagnosing TTP, the availability of assay is limited to special lab and it takes a lot of time for diagnosis. Hence, it is not reasonable to depend on ADAMT 13 assay in the diagnosis of TTP, but the diagnosis based on clinical circumferences is more judicious not to delay the treatment3,4). A recent targeted therapy with rituximab has been tried to alleviate the excessive immunologic responses in TTP, the results is currently anecdotal. Thus, therapeutic plasmapheresis (TPE) has been played a pivotal role in the treatment of TTP5,6). In fact, prior to plasmapheresis was applied to treatment, the mortality rate of TTP had been reached 90%, but it decreased to less than 20% after introduction of plasmapheresis7). In this report, clinical vignette of postoperative CNS-involved TTP treated by plasmapheresis and related literature would be reviewed.
CASE
A 78-year-old woman with the history of cerebral infarction three years ago, visited hospital because of right hip pain after slip down episode. In physical and radiologic examination, right hip fracture was diagnosed. There were no noticeable abnormalities in blood chemistry, cardiopulmonary and neurologic examination. Consequently, she underwent bipolar hemiarthroplasty (BPHA).
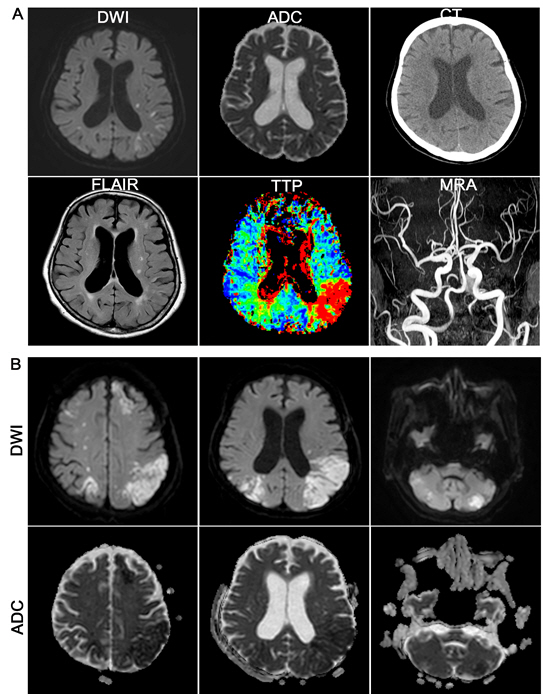
After 5-day recovery, she presented slightly decreased consciousness and disorientation without prominent neurologic deficits. In diffusion-weighted MR images, no diffusionŌĆōrestricted lesion revealed, however, cerebral blood flow delay was noted in time-to-peak (TTP) images (Fig. 1A). Unfortunately, global aphasia and hypobulic compliance were immediately followed by period of consistent deterioration of consciousness. Multifocal cerebral infarction was noted over frontal, temporal lobe and cerebellar hemisphere in diffusion-weighted image and ADC map (Fig. 1B). However, there were no evidence of thromboembolic source. Cerebral vasculature including carotid artery was patent without luminal irregularities in contrast-enhanced MR angiography. Additionally, cardiac arrhythmia had not been reported during hospital course in continuous EKG monitoring. At presentation, her platelet count was 13 ├Ś 109/L with mild normochromic normocytic anemia (hemoglobin was 10.3 g/dL). Initial blood smear showed occasional schistocytes with true thrombocytopenia (Fig. 2). On the following day, unexplained fever was developed and thrombocytopenia steadily worsened to 2.5 x 109/L. At same time, oliguria and blood pressure drop was sustained and revealed due to non-territorial cardiac dysfunction in echocardiography (Fig. 3). Cumulatively, the diagnosis of thrombotic thrombocytopenic purpura (TTP) was considered. She continued to deteriorate further with severe respiratory distress and did not improve despite intensive respiratory and circulatory supports including inotropic agents. The schistocyte count (based on classical triangular hyperchromatic RBCs) was slightly increased after a serial investigation. Direct antiglobulin test (DAT) was negative. Serial blood investigations noted worsening of anemia (8.4 g/dL) and thrombocytopenia (11 ├Ś 109/L) with reticulocytosis (7.5%). Lactate dehydrogenase (LDH) was 929 U/L (high) and liver function test (LFT) was abnormal. AST, ALT, and total bilirubin were 95 U/L, 99 U/L, and 32 umol/L, respectively. Results of coagulation screening tests (PT, APTT, and fibrinogen) were normal. D-dimer was elevated (3.68 ug/mL). Autoimmune profiles were negative for antinuclear antibody (ANA) and double stranded DNA (dsDNA). Under impression of TTP, therapeutic plasmapheresis was performed daily for 10 days with goal of normalization of platelet count and LDH.
Serial peripheral blood smear showed a reduced number of schistocytes in the blood film with increasing platelet count after plasmapheresis. Her condition was steadily improved, and she was transferred to general ward after 20 days of ICU admission following sustained normalization of platelet count, hemoglobin, and LDH levels (Fig. 4).
DISCUSSION
Pathogenesis
TTP is classified into congenital and acquired TTP. The congenital TTP accounts for less than 5% of total TTP and is usually presented at birth or in childhood. Congenital TTP occurs in an autosomal recessive manner by the homozygous mutation of the ADAMTS 13 gene located in the chromosome 9q34 locus. Most TTP is the form of acquired TTP. The von Willebrand factor (vWF), a glycoprotein secreted from vascular endothelial cells as a very large polymeric form, induces platelet adhesion and aggregation in vascular lesions. The ADAMTS 13, one of the metalloproteinases presents in plasma, cleaves the shear-induced conformational unfolded vWF. By clearing vWF before fully activating vWF formation by shear stress, ADAMTS 13 inhibits excessive vWF-mediated platelet aggregation1,4). Autoimmune TTP is caused by the production of anti-ADAMTS 13 antibody, which inhibits the proteolytic activity of ADAMTS 13. The anti-ADAMTS 13 antibody is usually an IgG type, binds to ADAMTS 13 and accelerates its removal from the plasma by opsonization or by an unknown mechanism. Because of ADAMTS 13 deficiency, uncontrolled large vVF polymer is formed and acts as a pivotal player in the manifestation of TTP clinical symptoms. Although the ADAMTS 13 deficiency is not consistently observed in all TTP patients, this is currently considered a model to elucidate the mechanisms of TTP2,5).
There have been known several predisposing factors that cause TTP as well as ADAMTS 13 deficiency (Table 1)4,8). Vascular endothelial damage is another central factor leading to thrombotic microangiopathy and could be caused by drugs, infections, toxins, and other clinical conditions that cause vascular endothelial damage. The patient in this report presented renal impairment and neurologic abnormalities after one week of hip hemiarthroplasty (BPHA). Although the mechanism how orthopedic surgery induces TTP is uncertain, it is not uncommon for TTP to occur after orthopedic surgery. There was no drug history that could induce the TTP except for orthopedic surgery.
Diagnosis
Paradoxically, thrombotic and thrombocytopenic-associated symptoms are coincidentally occurred in TTP9,10). Organ involvement by thrombosis in microvasculature manifests symptoms such as renal impairment, cardiac ischemia, central neurologic deficit. On the contrary, the hemorrhage symptoms including epistaxis, bruising, petechia, hematuria, hemoptysis, etc. are resulted from secondary thrombocytopenia caused by consumptive thrombocytosis. Moreover, non-specific symptoms such as fever, pallor, jaundice, fatigue, myalgia are also observed in TTP. In this case, ambiguous fever, renal impairment, cardiac dysfunction, neurologic deficits, hemorrhagic complications were accompanied. Characteristically, cardiac ischemia was presented as non-territorial pattern associated with multi-focal embolism in small coronary vessels9,11). Multifocal cerebral infarction caused by small vessel occlusion was also observed in diffusion weighted images. Given that there were no evidences of atrial fibrillation and carotid artery thrombosis, the source of microembolism was not evident.
In the laboratory test, the compatible findings for MAHA were observed and abundant schistocytes were revealed in peripheral blood smear. Direct antiglobulin test was negative. Severe thrombocytopenia, platelet count below 20,000/ul and mildly prolongated prothrombin time (PT) within 5 seconds of upper limit were also revealed. These are reasonable findings indicating TTP rather than DIC. The viral antigen and autoantibody test were also negative1,2,4).
Recent studies have recommended measuring the ADAMTS 13 activity before treatment. The ADAMTS 13 assay includes cleaving activity of ADAMTS 13, neutralizing or non-neutralizing autoantibody measurement. The severely decreased ADAMTS 13 activity (<5%) or the presence of IgG antibody to ADAMTS 13 establish the diagnosis of TTP. In case of severe ADAMTS 13 activity (<5%), the diagnostic specificity for TTP reaches to 90%. However, there are limitations in the ADAMTS 13 assay-guided treatment of TTP because the low sensitivity (60%), low accessibility of tests and lagging time to report the results. Patients in this case were diagnosed as TTP based on clinical and laboratory findings prior to confirmation of ADAMTS 13 assay4,10,11).
Treatment
Two-thirds of cases of TTP patients are sporadic TTP, and one-third of patients tend to relapse after TTP remission. Chronic recurrent form of TTP may be associated with genetic predisposition and autoantibody. On the other hand, in cases which link to malignancy or transplantation, they are usually presented as acute episode.
In the past, therapeutic splenectomy was performed in patients with refractory or chronic recurrent TTP. Currently, there are better treatment options than splenectomy in the aspect of surgical risk-benefit, but the splenectomy had been shown a firm effect to prevent recurrence of TTP. There were retrospective studies showed the remission rate of the splenectomy after TTP episode. When observing average 111 months (range, 9 to 230 months), relapse-free survival reached 10 years and relapse rate was 17% when splenectomy was done. Currently one of the most important treatment methods of TTP is daily plasmapheresis and it has been reported that the mortality rate is decreased by about 90 to 20%. It restores ADAMTS13 by removing autoantibody from the blood8,12,13). Recently, early plasmapheresis is recommended because in cases that plasmapheresis is delayed, usually need a large volume of plasma infusion. The total period and number of plasmapheresis required for TTP remission are highly variable depending on the patients, however, obviously long period of plasmapheresis is needed especially in the antibody-mediated TTP. Daily exchanges should last at least two days even after reaching the normal level of platelet count (> 150 ├Ś 109/l). Transfusion is one of the essential adjunctive therapies during therapeutic plasmapheresis. Red cell transfusion should be administered according to clinical need, especially when anemia is getting worse. During active hemolysis, folate supplementation could be also required. Unless TTP is associated with life-threatening hemorrhage, platelet transfusion is not routinely indicated. Prophylactic use of LMWH to prevent thrombosis in high risk patients is recommended once the Platelet count reaches above 50 ├Ś 109/l.
Steroid is widely used during plasmapheresis at early period of acute immune TTP. High dose steroids were reported to show minimal side effects and improve patient prognosis. However, there is no randomized clinical trial proving that a combination of plasmapheresis and steroid is superior to plasmapheresis alone5,10,13).
Relapse
Relapse is defined by TTP recurrence after 30 days from initial episode. The TTP relapse is known to occur in 20 to 50% of total TTP cases. Canadian Apheresis group reported 36% relapse rate over a 10 year follow up of TTP patients.
Patients with ADAMTS 13 activity less than 10% or in which anti-ADAMTS 13 antibody is positive in blood during remission period are reported to show a 3-fold higher recurrence rate over 1 year. In case that ADAMTS 13 activity was 5% or less, the recurrence rate reached 38.5%, but in case of 15% or more, only 5% of the patients relapsed in previous report4,5,12).
The use of rituximab during acute TTP episode was reported to reduce the risk of relapse. In patients with ADAMTS 13 activity less than 5%, elective rituximab therapy could normalize ADAMTS 13 activity and decreased relapse rate. Cumulatively, TTP patients are recommended to counsel the symptoms and dangers of recurrence and monitor ADAMTS 13 activity periodically.